細胞治療:科學監管如何引導產品開發良性發展?
本文授權轉載自微信公眾號:醫麥克
2019年3月29日,國家衛健委發布關于征求《體細胞治療臨床研究和轉化應用管理辦法(試行)(征求意見稿)》,引起行業內眾多從業者的巨大反響。這一幕也大概出現在一個月前(2月26日),同樣由國家衛健委發布了《生物醫學新技術臨床應用管理條例(征求意見稿)》。兩份監管文件主要關注的焦點就是細胞治療,或者更具體說為體細胞治療。
在魏則西事件之后,體細胞治療的臨床收費應用被叫停,統一納入臨床研究范疇進行規范管理。隨后衛計委和CFDA聯合發布了《干細胞臨床研究管理辦法》,后續緊跟著又出臺了《干細胞制劑質量控制和臨床前研究指導原則》,依據該辦法開展干細胞臨床研究后,如申請藥品注冊臨床試驗,可將已獲得的臨床研究結果作為技術性申報資料提交并用于藥品評價。再之后2016年食藥監總局出臺了《細胞制劑研究與評價技術指導原則》征求意見稿,并于2017年12月調整為《細胞治療產品研究與評價技術指導原則(試行)》發布。之后隨著南京傳奇獲得個IND受理和批件之后,國內有20多家企業按照藥品的原則進行了CAR-T細胞制品的IND申報。
細胞治療按照醫療技術還是藥品來管理,世界各國有所不同,這是各國管理體系的不同帶來的。隨著2017年美國FDA專家咨詢委員會以10:0的票數通過首個CAR-T療法后,為細胞治療按照藥品監管鋪平了道路。然而,考慮到細胞治療產品的特異性,在體細胞治療的監管方面存在諸多的分歧,例如CAR-T細胞治療臨床研究與臨床應用按照藥品還是技術監管,臨床研究的監管主體和審評標準,醫療機構進行臨床應用如何與藥品GCP原則一致,倫理的監管原則與是否患者受益化等。這些問題目前在出臺的法規條例中還沒有更詳細清晰的解釋。
以下我們將對監管較為領先的美國、歐盟與日本進行闡述,希望能從中獲得啟發。
產品概念界定
美國將細胞、組織或基于細胞、組織的產品(HCT/Ps)歸類監管,歐盟以先進技術治療醫學產品(advanced therapy medicinal product,AP)歸類監管,日本則按照再生醫學產品管理。
美國、歐盟、日本對于細胞治療產品的概念界定的側重點各有不同,美國注重界定制藥方式;歐盟側重于臨床應用范圍,可用于疾病的預防、診斷或治療,并且強調了被處理和生物學特性;日本則強調了細胞的來源為自體或同源,對細胞進行的人工基因操作技術和主要用于治療與再生修復。因此可以歸納出,細胞治療的產品界定可從細胞的來源、細胞人工操作的技術范圍、制藥方式、臨床適用的范圍進行約束。

(圖片來源:《中國醫藥生物技術》)
細胞治療審批監管法規政策框架
雖然不同國家衛生與藥監管理體系職能有所不同,從監管與審批來看細胞治療產品與臨床應用總體分為兩條路徑:
一是以藥物或醫療器械產品由藥品監管部門進行臨床準入與應用的監管審批,需要嚴格遵循藥物產品審批的流程;
二是醫療技術由衛生部門進行監管,在醫院直接進行臨床應用,不同國家法規界定的細胞治療類型與應用有所差異。
1
日本
日本的“再生”熱潮基于一系列藥品改革和新法律,尤其是具有里程碑意義的《藥品和醫療器械法》(PMD法案)和《再生醫學安全法》,能夠為再生提供有條件的營銷批準,因此可以實現更快地商業化,這是一個被廣泛認為是世界上最快的審批流程。
日本將細胞治療、基因治療、組織工程作為獨立于藥物、醫療器械的再生醫學產品單獨監管,并在 2013年進行了再生醫學產品的審批改革。2013年日本出臺了關于細胞制品的管理規范,將細胞類產品歸類為新分類:再生醫療等制品。該管理辦法規定:對于均質性不一的再生醫療等制品,如果能確定其安全性,并且能估計其有效性,那么可以通過附加條件及期限,特別是在早期就可以對其予以承認。然后,再重新驗證其安全性和有效性。這個政策的變化令個性化醫療的管理再次向前邁進了一大步。
再生醫療領域的主要國家監管部委為:厚生勞動省、經濟產業省、文部科學省、藥品和醫療器械管理局(PMDA)。四個機關單位在研究推動、設計開發、許可認定、品質評價、程序審查等具體事務上各有側重和分工協作,另外日本規格協會(JSA)負責安全性評價等行業標準制定。
再生醫療創新論壇(FIRM)、日本再生醫療學會、京都大學iPS細胞研究所(CiRA)等主要相關機構也在細胞采集、制備、運輸、保存等具體技術層面上都制定了業內指導文件。
2010年以來,經濟產業省主要對細胞培養相關設備及工藝流程等方面制定了一些指導方針。近年,厚生勞動省主導制定的法規較多,針對不同的疾病領域、細胞類型等都分門別類地做了詳細規定,也是評估審查的主要參照標準。
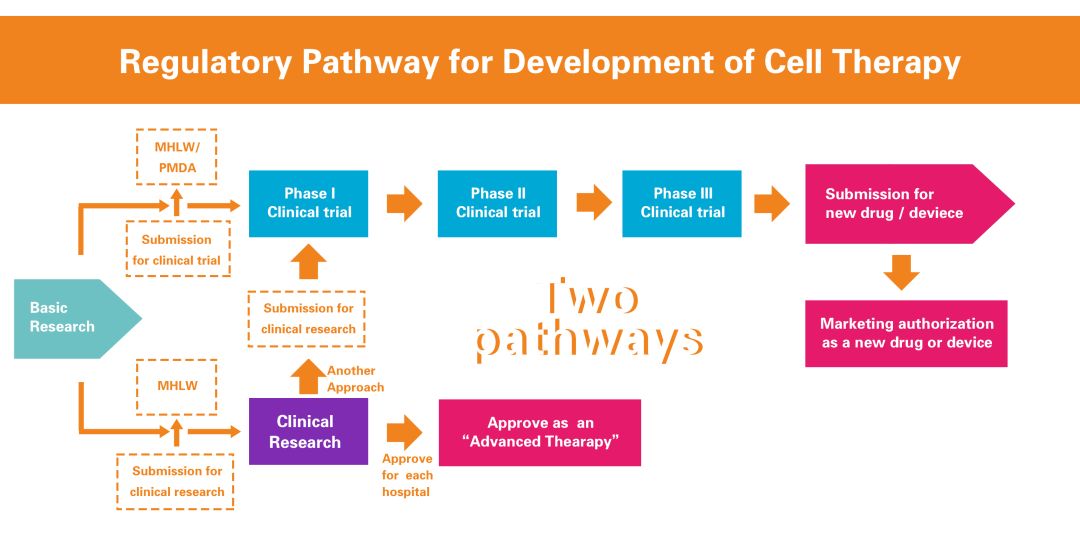
日本的研究性醫療產品的臨床開發有2種途徑(雙軌制):注冊試驗和臨床研究。注冊試驗是指以上市許可為目的進行的臨床試驗。臨床研究不用于上市許可。注冊試驗不僅由公司進行,而且由學術研究人員作為研究者發起的注冊試驗進行。在臨床研究中,醫生給病人施用醫療產品作為研究。注冊試驗和臨床研究受到不同的監管。
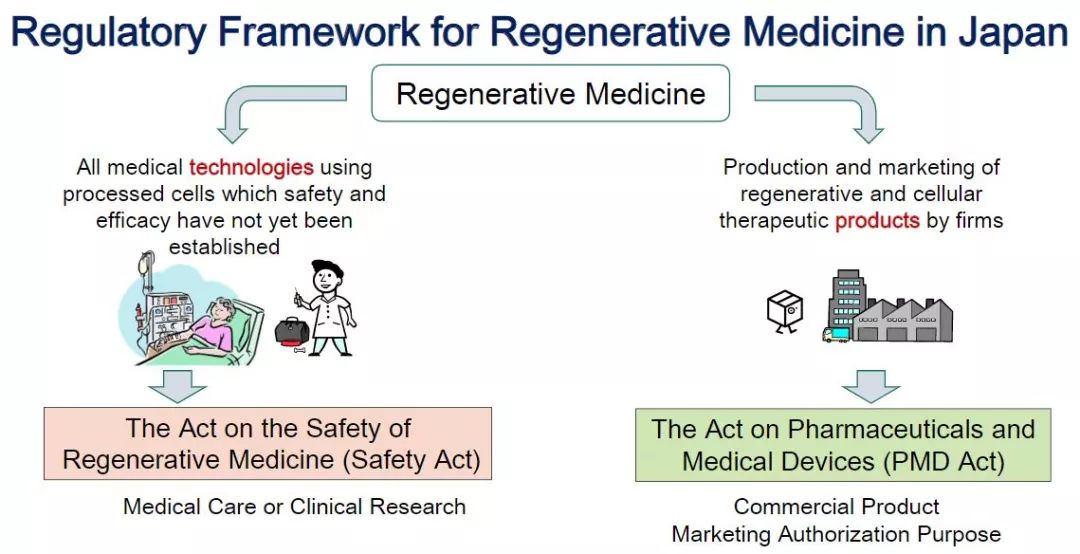
日本再生醫學監管框架(圖片來源:PMDA、iGEM 2016)
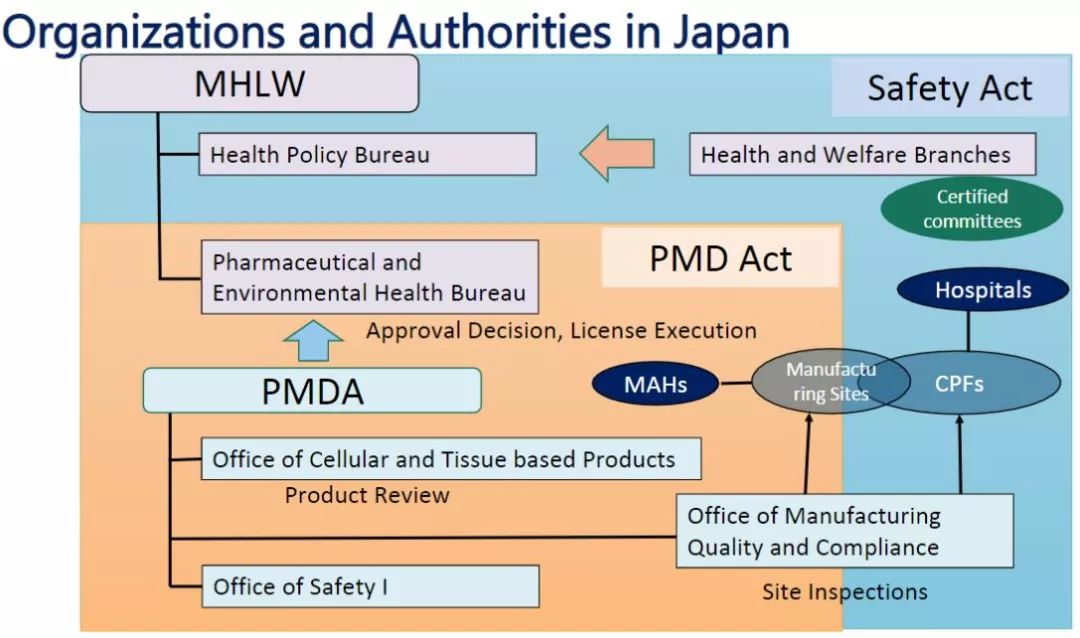
組織和管理機構(圖片來源:PMDA)
在注冊試驗和上市許可方面,藥品和醫療器械管理局(PMDA)審查注冊試驗和上市許可申請的研究性新藥通知,并且厚生勞動省(MHLW)批準藥品、醫療器械和再生醫學產品的上市許可。在臨床研究方面,MHLW主要負責監管,PMDA僅負責細胞加工設施的調查。

(圖片來源:《中國醫藥生物技術》)
(一)注冊試驗
2014年11月,日本厚生勞動省(MHLW)修訂了舊的藥事法,并實施了新的《藥品和醫療器械法》(PMD法案),適用于上市許可的注冊試驗。在該法案中,細胞醫療產品、離體基因醫療產品和體內基因醫療產品被新定義為“再生醫學產品”。
再生醫學產品由PMDA依據《藥品和醫療器械法》進行監管,其藥品評估中心下設細胞與組織類產品審批辦公室負責具體審批事務。再生醫學產品在原有藥物審批程序基礎研究、臨床研究、臨床試驗、審批準入的基礎上,在臨床研究證實再生醫學產品的有效性與安全性之后,增加了條件性限制性準入許可。
該批準系統類似于美國的加速審批系統和歐盟的有條件的審批系統(僅適用于初始上市許可)。再生醫學產品需要滿足以下條件:適應證為危及生命的疾病,治療方法為滿足需求的創新性產品,并經過初步的有效性、安全性驗證,符合相關監管法規政策。在經過患者知情同意后產品進入市場,大大加快了產品臨床應用的進程。
條件性限制性準入許可時間最長為 7 年,在證明細胞治療產品臨床試驗與應用有效性之后,產品可以申請作為正式的再生醫學產品長期上市,7 年時間到期后再次進行申請或者退出市場。目前已有一種用于缺血性心臟病嚴重心衰的骨骼肌細胞產品通過條件性限制性準入許可進入市場,市場考察期為 5 年。
此外,MHLW于2015年4月設立了SAKIGAKE(意為日語的先驅者或先行者)藥物指定系統。2015年7月,MHLW另外啟動了醫療器械、體外診斷和再生醫學產品的SAKIGAKE指定系統。
SAKIGAKE指定:
要求
1. 行動方式是創新的
2. 適應癥嚴重
3. 預期效果顯著
4. 醫療產品在日本開發,發起者計劃首先在日本提交上市許可申請
好處
1. 優先咨詢(減少等待時間)
2. 大量預申請咨詢
3. 優先審查
4. 指派PMDA管理者作為禮賓人員
SAKIGAKE指定的藥物和醫療器械的目標總審查時間為6個月(不適用于SAKIGAKE指定的再生醫藥產品)。一般而言,基因和細胞產品的目標總審查時間為9個月;審查時間不包括公司準備對PMDA的查詢作出答復的時間。此SAKIGAKE指定類似于美國的突破性治療指定和歐盟的PRIME(優先藥物)。

早期審評推進計劃(圖片來源:PMDA)
此外,MHLW于2012年啟動了“創新藥物、醫療器械和再生醫療產品實際應用項目(Project for Enhanced Practical Application of Innovative Drugs, Medical Devices and Regenerative Medical Products)”,以促進PMDA與學術機構合作,共同起草創新醫療產品評價的監管指導文件。正如MHLW計劃的那樣,該項目于2017年3月31日完成。
(二)臨床研究
2014年11月,MHLW發布了《再生醫學安全法》(ASRM),適用于使用細胞醫療產品進行注冊試驗以外的臨床研究。因此,體內基因治療不在ASRM的范圍內,盡管PMD法案中定義的再生醫學產品包括體內基因治療。離體基因治療,如采用基因轉移的過繼免疫治療,則屬于ASRM的范圍。
根據該法案,由經認證的委員會審查醫療或臨床研究計劃,并強制向厚生勞動省(MHLW)提交計劃。
在涉及體內基因治療的臨床研究中,醫生必須遵循“使用體內基因治療的臨床研究指南(the Guideline on Clinical Research Using In Vivo Gene Therapy)”。一些(不是全部)在日本進行體內或離體基因治療的注冊試驗和臨床研究列于國立衛生科學研究所的網站上。
在ASRM的監管下,使用加工細胞的臨床研究分為I類(高風險),II類(中等風險)或III類(低風險)。胚胎干細胞、誘導多能干細胞、遺傳修飾細胞、動物細胞或同種異體人細胞的使用被歸類為I類;將自體細胞施用于與施用細胞相似的器官(同源使用)被歸類為III類;將自體細胞用于其他目的的用途分類為II類。
目前,任何涉及離體基因轉移的I級和II級醫學治療或臨床研究均需經大阪大學再生醫學認證特別委員會審查。該委員會只能審查在任何日本醫院進行的涉及離體基因轉移的臨床研究的應用。
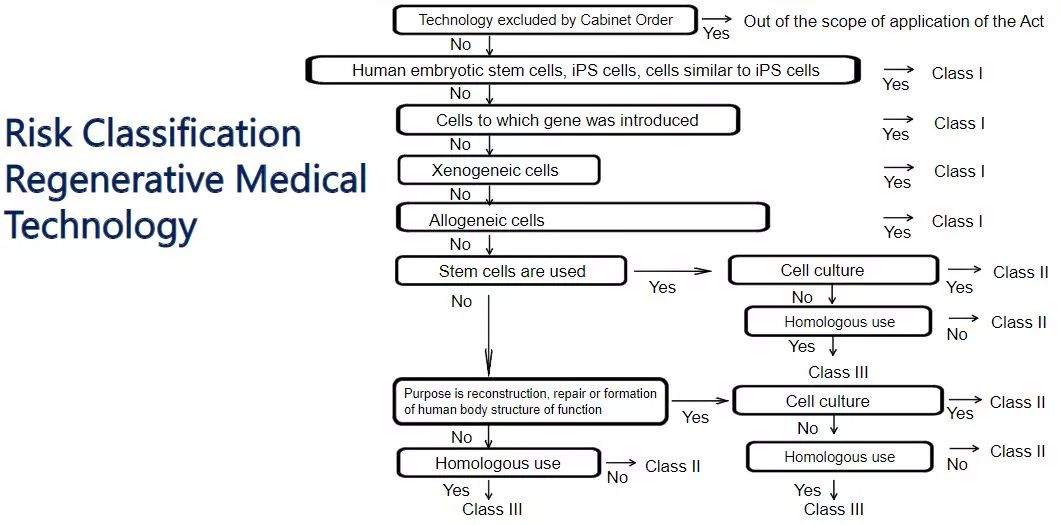
再生醫療技術風險分類(圖片來源:PMDA)
《臨床試驗法》自2018年4月起生效,該法規定了進行“特定臨床試驗”的程序、認證審查委員會適當運作的措施,以及關于臨床試驗資金或其他利益信息的公布。特定臨床試驗分為以下幾類:接受公司提供的研究資金或其他利益的臨床試驗;對未經批準的藥品、醫療器械、再生醫學產品,以及經批準的藥品、醫療器械、再生醫學產品以未經批準的適應癥、劑量進行的臨床試驗。ASRM將進行修訂,以確保與《臨床試驗法》保持一致。
總體來講,如果僅是在診所或醫院等機構內部實施的免疫細胞采集和治療,以及研究者發起的臨床試驗,屬于《再生醫學安全法》的管轄范疇。如果有第三方企業參與免疫細胞的基因操作、加工制備、生產銷售等,則歸《藥品和醫療器械法》(修訂后的《藥事法》)管轄。
2
歐盟
歐盟將組織工程、細胞治療、基因治療產品納入先進技術治療醫學產品(AP)管理,該類藥物的定義是能夠為疾病帶來革命性的治療方案,對于患者與產業具有巨大前景。
歐盟細胞治療的監管有兩條路徑:一是按照先進技術治療醫學產品進行臨床研究與申報,由歐洲藥品管理局(EMA)負責審批和管理;二是遵循醫院豁免條款,由醫院決定對患者的治療應用。
從法律層面,細胞治療管理的法律依據為歐盟《醫藥產品法》與《醫療器械法》,對醫藥產品的臨床前研究、臨床研究、制造與銷售進行全產業鏈的系統提供法律監管框架。
隨著生物治療的飛速發展,歐盟加強了對細胞治療產品的監管,2007 年歐盟頒布了《先進技術治療醫學產品法規》(Regulation (EC) No. 1397/2007 on Advanced Therapy Medicinal Product),于2008年12月30日起實施,將基因治療產品、體細胞治療產品和組織工程產品定義為先進技術治療醫學產品,其中細胞治療產品指含有經過處理的被改變了生物學特性的細胞或者組織,可以用于疾病的治療、診斷或者預防。按照藥物申報,先進技術治療醫學委員會審批,審批時間1~2年。該法規中提出了醫院豁免條款(Article 28),對某一醫生進行的,為患者個體進行的治療應用行為進行豁免。該條款允許歐洲醫院在經過基礎研究、臨床研究驗證有效性與安全性之后,可以生產小規模的細胞產品用于特定的患者,主要是臨床中心進行自體細胞治療。

(圖片來源:《中國醫藥生物技術》)
此外,歐盟針對基因治療和細胞治療產品還制定了一系列科學指導原則。這些指導原則提出了對 AP 的研發和監管要求,如基于風險的產品開發途徑和評價理念、對于細胞和結構組分之間相互作用的特殊要求、對于臨床/非臨床的靈活性考慮、對于藥品臨床試驗管理規范(GCP)的特殊要求,以及關于上市后安全有效性跟蹤和風險管理的特殊考慮等。
在審評程序方面,歐盟規定AP必須執行集中化審評程序,并成立了先進技術療法委員會(CAT),專門負責新技術療法產品的技術審評。CAT對每一份提交至管理部門的AP提出審評意見,但該意見將被提交至人用醫藥產品委員會(CHMP),由CHMP做出采納批準、變更、暫停或取消上市許可的建議,然后將建議發送至歐盟委員會做決定。一旦產品在歐盟被批準上市,管理部門將對其安全性和有效性進行進一步評價。同時,為鼓勵 AP 的研究和開發,歐盟執行了一些特殊的支持鼓勵政策,如減免申請人向管理部門支付的部分費用、申請人可從歐盟獲得更多科學支持和幫助等。
2016年3月,歐洲藥品管理局(EMA)推出了名為優先藥物(PRIME)的快速審批方案,獲準進入PRIME計劃的實驗性藥物,將在臨床試驗及醫藥開發方面獲得EMA大量支持,加速真正創新藥物的開發的審批,以滿足對有前景新藥的醫療需求。
3
美國
美國在細胞治療領域已經形成了完善的法規監管框架,由上位法律、法規、管理制度與指南三層組成其法律法規體系。美國細胞、組織或基于細胞、組織的產品(human cells, tissues, or cellular or tissue-based products, HCT/Ps)屬于人類組織和細胞類產品范疇 ,分為 PHS 351 產品與PHS 361 產品兩大類管理,PHS351 產品由 FDA的生物制品評估研究中心(CBER)統一負責審批,PHS 361 產品可以在醫院直接進行臨床應用。
法律法規從法律層面,細胞治療管理的法律依據來自于兩個國會法案,即《美國食品、藥品和化妝品法案》(FD&C Act)及《公共衛生服務法案》(PHS Act)。《美國聯邦條例》(CFR)是 FDA管理要求的構建基礎,最適用于干細胞的法規是對提交研究性新藥申請要求做出的規定(21CFR312),以及關于現行藥品生產質量管理規范的法規(21CFR 210&211)。《現行藥品生產質量管理規范》(cGMP)是根據 CFR 中的 21CFR 210 和 211 條制定的,其中的原則適用于干細胞產品,包括制造產品設備的物理特征及在某種設備中制造細胞產品的過程和步驟。
美國于 2001 年發布 CFR1271 管理法規,并于 2005 年正式實施。這是細胞治療審批主要依據的法規,將人體細胞組織分為 PHS 351 產品與PHS 361 產品兩大類管理,PHS 351 產品屬于HCT/Ps 分類監管的產品,包括骨、韌帶、皮膚、硬腦膜、心臟瓣膜、角膜、外周血干細胞(PBSCs)、臍帶血來源前體細胞、經過改造的自體軟骨細胞、人工合成基質上的表皮細胞、精子或其他生殖組織。美國已經獲批的CAR-T產品正是按照PHS 351 產品進行監管的。
指南與規范 FDA 還與其他細胞治療領域管理部門、企業、研究機構相互溝通、相互影響,由此形成了一些有關大多數種類生物產品的制造和臨床試驗的指南規范。在這些共同制定的大量指導性文件中,界定的原則適用于細胞療法的評估。FDA 和 NIH 之間通過簽署正式的諒解備忘錄(MOU)協議促進干細胞管理建議的形成。
審批監管框架 FDA 生物制品評估研究中心下設細胞、組織與基因治療辦公室,該辦公室由人類組織管理、臨床評估與藥理、細胞與基因治療三個部門組成,其中細胞與基因治療部負責接收細胞治療產品的審批與準入,快速審批程序時間為6~10 個月,目前美國已有多種細胞治療產品上市。
PHS 361 類細胞治療產品無需向 FDA 提出IND 申請,不屬于 HCT/Ps 分類監管的產品,包括全血、血液成分或血液衍生產品,如白細胞、血小板、凝血因子等(由血液制品相關規定監管)動物來源的細胞、組織或器官干預最小化、作為同源性應用的骨髓等。
PHS 361 產品需要同時滿足以下條件:制備過程符合干預最小化,僅同源性使用,未添加水、晶體液或殺菌、保存、存儲劑之外的其他任何試劑。并且不產生全身反應,且不依賴活體細胞的代謝過程發揮作用;若產生全身反應或依賴活體細胞的代謝過程發揮作用,則必須是符合作用同源性且用于 1、2 級親屬的異基因移植或作為生殖應用。其中產品最小化干預(minimally manipulated)是指在細胞的處理過程中,不能改變相關的生理特性(即未經過體外激活、包裹、擴增或基因修飾等)。
4
中國
2003年3月,國家食品藥品監督管理局發布了《人體細胞治療研究和制劑質量控制技術指導原則》,要求每個方案的整個操作過程和最終制品必須制定并嚴格執行標準操作程序,以確保體細胞治療的安全、有效。
2007年,國家發改委、衛生部、國家中醫藥管理局聯合印發了《全國醫療服務價格項目規范》,對樹突狀治療(DC)及LAK細胞治療進行了費用規定。2012年對規范進行了修訂,對腫瘤免疫治療的診療服務價格進行了統一規定,為醫藥費用的收取提供了法律依據。在醫療保障方面,根據衛生部新增的醫療項目,DK-CIK生物治療已納入國家醫保項目范圍,省醫保可報銷90%,市醫保可報銷80%。這是細胞治療被用于廣大民眾的一個利民舉措。
2009年3月,衛生部制定印發《醫療技術臨床應用管理辦法》(衛醫政發〔2009〕18號)規定第三類醫療技術由衛生部負責技術審定和臨床應用管理。研究機構證實動物試驗和臨床試驗有效,提交申請給衛生部,經衛生部審定批準后再用于臨床治療。
2009年5月,衛生部發布《首批允許臨床應用的第三類醫療技術目錄》,將自體免疫細胞(T細胞、NK細胞)治療技術歸位第三類醫療技術。首次把細胞治療技術作為第三類醫療技術,從此自體免疫細胞(T細胞、NK細胞)治療技術有了更加明確的管理部門。
2009年6月,衛生部為規范自體免疫細胞(T細胞、NK細胞)治療技術臨床應用,保證醫療質量和醫療安全,制定了《自體免疫細胞(T細胞、NK細胞)治療技術管理規范(征求意見稿)》。對自體免疫細胞(T細胞、NK細胞)治療技術制定了管理規范,這使得該項技術有了可尋的質量管理細則。
2010年,衛生部審議通過了《藥品生產質量管理規范》(2010年修訂)》,貫徹質量風險管理和藥品生產全過程管理的理念,與世界衛生組織的GMP規范接軌,成為細胞在實驗室制備過程中必須遵循的規范。生產質量管理需要更加標準,與國際GMP規范接軌標志著我國的藥品生產將更加嚴格,實現從有到優的轉變。
2011年6月,衛生部公布第三類醫療技術審核機構名單,將中華醫學會、中國醫院協會、中國醫師協會、中華口腔醫學會作為第三類醫療技術審核機構,有效期為自2011年5月2日至2013年5月31日。審核機構的公布代表了對權威機構的認證。
2011年11月,國家“十二五”生物技術發展規劃(國科發社〔2011〕588號)明確發展重點包括:“針對惡性腫瘤、心腦血管疾病、遺傳性疾病、自身免疫性疾病等嚴重威脅人類健康的重大疾病,開展一批靶向基因治療、細胞治療、免疫治療等前瞻性的生物治療關鍵技術研究,以關鍵技術的突破來帶動重點產品的研發,加快生物治療技術應用于臨床治療的速度。”國家層面的政策支持,加快生物治療技術應用于臨床治療,對于國家技術創新和國民健康來說具有重大意義。
2011年12月,衛生部和國家食品藥品監督管理局聯合發布《關于開展干細胞臨床研究和應用自查自糾工作的通知》)(衛辦科教函【2011】1177號),決定聯合開展為期一年的干細胞臨床研究和應用等活動。自行開展、沒有經過任何審批的行為要立刻停止。對于已經經過食藥監局批準的干細胞制品的臨床試驗項目,要按照批件和藥品臨床試驗有關質量規范的要求嚴格執行,不能隨意變更臨床試驗方案,更不能收費。同時,在2012年7月1日之前,停止的新項目申報。行業亂象頻出,行業健康發展需要監管部門的介入。
2012年7月,科技部發布《“十二五”生物技術發展規劃》,把干細胞與再生醫學技術、基因治療與細胞治療技術列入發展重點。整頓之后的行業進入良性發展,列入發展重點代表了國家對于這些技術突破的期望。
2012年12月,國務院印發生物產業發展規劃的通知(國發〔2012〕65號)明確將抗腫瘤藥物、治療性疫苗、細胞治療等列為重要發展和重點支持的產業。技術的創新,推動產業的發展,重點扶持將使得技術產生的產品更快被民眾所用,這是技術落地的關鍵性政策。
2013年,國家衛計委及CFDA聯合發布《干細胞臨床試驗研究管理辦法(試行)》、《干細胞臨床試驗研究基地管理辦法(試行)》和《干細胞制劑質量控制和臨床前研究指導原則》(試行)》3個文件的征求意見稿,指出干細胞臨床試驗研究必須在干細胞臨床研究基地進行,研究基地必須具備三級甲等醫院和藥監局認定的藥物臨床試驗機構等要求。對于臨床試驗管理辦法、研究場所、認證機構進行規定,使得私自開展的臨床試驗無處遁形。
2015年7月,國家衛計委發布了國衛醫發[2015]71號《國家衛生計生委關于取消第三類醫療技術臨床應用準入審批有關工作的通知》中明確指出“取消第三類醫療技術臨床應用準入審批后,醫療機構對本機構醫療技術臨床應用和管理承擔主體責任。各級各類醫療機構應當按照《醫療技術臨床應用管理辦法》(衛醫政發[2009]18號)要求,強化主體責任意識,建立完善醫療技術臨床應用管理制度,按照手術分級管理要求對醫師進行手術授權并動態管理,建立健全醫療技術評估與管理檔案制度。”當行業的要求、共識建立起來,強化主體責任意識使得監管更加靈活,將更快加速行業發展。
2016年,可謂是細胞治療生死攸關的一年。5月魏則西事件之后,國家衛計委重申禁令,叫停了全國范圍內不同醫院正在開展的免疫細胞治療。5月4日,國家衛生計生委召開了關于規范醫療機構科室管理和醫療技術管理工作的電視電話會議,會議重申,自體免疫細胞治療技術按照臨床研究的相關規定執行。業界普遍認為,該規定進一步明確了國內免疫細胞治療的行業標準,規范了臨床應用,對推動整個行業的健康持續性發展具有重大意義。
2016年12月16日,發布《細胞制品研究與評價技術指導原則》(征求意見稿)以來,細胞治療政策落地反映國家對醫療創新技術的高度支持和鼓勵,將直接利好細胞治療技術公司。
2017年12月,CFDA發布了《細胞治療產品研究與評價技術指導原則(試行)》,規范和指導這類產品按照藥品管理規范進行研究、開發與評價,制定本指導原則。
2018年,12月13日,國家衛生健康委員會官網刊發了《關于政協十三屆全國委員會次會議第4443號(醫療體育類434號)提案答復的函》,至今已有102家醫療機構和19個臨床研究項目完成備案,使干細胞治療技術從基礎研究進入臨床研究,有力推動我國干細胞治療技術發展。借鑒干細胞臨床研究管理模式,組織開展細胞治療技術臨床研究機構申報、遴選、備案。全力支持細胞治療產品申報藥物注冊。
生物醫學新技術臨床應用管理條例
(一)明確了管理范疇。科技部負責起草《生物技術研究開發安全管理條例》,規范生物技術的基礎研究。本條例規范生物技術的臨床階段研究和轉化應用。據此,將生物醫學新技術定義為:已完成臨床前研究,擬作用于細胞、分子水平的,以對疾病作出判斷或預防疾病、消除疾病、緩解病情、減輕痛苦、改善功能、延長生命、幫助恢復健康等為目的的醫學專業手段和措施。
(二)建立了生物醫學新技術臨床研究和轉化應用行政審批制度。一是規定醫療機構開展生物醫學新技術臨床研究和轉化應用必須經過行政部門批準。二是規定了開展生物醫學新技術臨床研究醫療機構和項目主要負責人的條件。三是明確衛生行政部門審批以學術審查和倫理審查為基礎。四是對生物醫學新技術的臨床研究按照風險等級進行兩級管理,中低風險研究項目由省級衛生主管部門審批,高風險研究項目由省級衛生主管部門審核后國務院衛生主管部門審批;研究成果轉化應用均由國務院衛生主管部門負責。
(三)規定了學術審查和倫理審查的主要內容。借鑒國際和世界衛生組織倫理審查有關規定,條例規定了衛生主管部門進行學術審查和倫理審查的主要內容,增強審查嚴肅性和規范性。同時規定審查規范,包括倫理委員會、學術委員會組成,審查具體技術規范,審查結論等另行制定。
(四)強調機構主體責任。明確開展(包括牽頭或參與)臨床研究的醫療機構承擔主體責任。明確開展臨床研究的醫療機構應當具備一定的條件,具體條件另行制訂。醫療機構主要負責人是本機構臨床研究管理的責任人。醫療機構為其他機構提供技術支持、研究場所,提供人體細胞、組織、器官等樣本,協助進行志愿者招募的,本機構及參與人員同樣承擔相應責任。
(五)加大了違規處罰力度。針對現有規定處罰力度弱,無法形成威懾的問題,條例加大了違規行為的處罰力度。對醫療機構違規開展臨床研究和轉化應用、未按規定開展研究、醫師違反規定、其他醫務人員違反規定、非醫療機構違規開展臨床研究等情形明確了處罰措施,包括警告、限期改正、罰款、取消診療科目、吊銷《醫療機構執業許可證》,開除或辭退,終生不得從事生物醫學新技術臨床研究等;情節嚴重的還將追究刑事責任。
(六)與藥品和醫療器械管理進行銜接。部分生物醫學新技術臨床研究的預期成果為藥品或醫療器械,條例規定按照《藥品管理法》和《醫療器械監督管理條例》等規定管理。
體細胞治療臨床研究和轉化應用管理辦法(試行)解讀:
體細胞的科學定義包括干細胞,原國家衛生計生委會同原食品藥品監管總局于2015年出臺了《干細胞臨床研究管理辦法(試行)》,有效地規范和推動了我國干細胞治療臨床研究工作,按照本管理辦法“第三十一條,有專門管理辦法的體細胞治療按照現行規定執行”,干細胞臨床研究等有關工作仍然按照《干細胞臨床研究管理辦法(試行)》的規定執行。
本管理辦法第三條規定“本辦法適用于由醫療機構研發、制備并在本醫療機構內開展的體細胞治療臨床研究和轉化應用”。國家衛生健康委對細胞治療轉化應用項目進行目錄管理,與產業化前景明顯的細胞治療產品錯位發展。由企業主導研發的體細胞治療產品應當按照藥品管理有關規定向國家藥品監管部門申報注冊上市。
按照本管理辦法有關規定,開展體細胞治療臨床研究和轉化應用的醫療機構應當進行備案,醫療機構進行首次機構備案時,須同時提供醫療機構備案材料和臨床研究項目備案材料。
體細胞治療臨床研究不得向受試者收取任何研究相關費用。體細胞治療轉化應用項目備案后可以轉入臨床應用,由申請備案的醫療機構按照國家發展改革委等4部門《關于印發推進醫療服務價格改革意見的通知》(發改價格〔2016〕1431號)有關要求,向當地省級價格主管部門提出收費申請。
5
總結
上述是多國細胞治療的監管情況,確實有不同的地方。監管原則和方法是否可以大膽借鑒國際上的一些優點并結合我們自身的實踐完善監管體系。從2017年年底開始,陸續有CAR-T細胞、TCR-T細胞、干細胞、DC疫苗按照藥品的申報和審評獲得受理和批件,這條剛剛開始的創新審評之路在發展過程中是否還存在待需完善的機制,是否可以結合藥監部門對藥品注冊審評的經驗和醫療機構良好的臨床研究和倫理經驗進行優勢整合,從而保證不同來源的細胞制品都有非常好的安全性和療效評估保障。
當然我們有自己特殊的國情,所以非常理解與支持從嚴監管的目標與準則。希望多聽取各方建議,科學嚴謹但不失創新的態度繼續為推動中國細胞治療產業健康發展共同努力。
參考出處:
美國、歐盟、日本細胞治療監管政策研究, 《中國醫藥生物技術》2016年12月第11卷第6期
https://www.pmda.go.jp/files/000219466.pdf
客觀日本:細胞治療與再生醫療,中日監管大不同(上)(下)
https://cdn.ymaws.com/www.celltherapysociety.org/resource/resmgr/2015_AnnualMtgPresentations/GRP_Session_1_Akihiro_Shimos.pdf
https://www.clinicaltherapeutics.com/article/S0149-2918(18)30552-6/fulltext
https://www.bsigroup.com/zh-CN/medical-devices/market-access/japan/