
УГ‘фОДХВЈәҪYәПЩ|ЧVө°°ЧЩ|ңyРтҪвҙaө°°ЧЩ|МЗ»щ»Ҝ
МЗө°°ЧХјИЛоҗө°°ЧЩ|әНҙу¶а”өЙъОпЦЖЛҺ®aОпөД50%ТФЙПЈ¬ө°°ЧЩ|МЗ»щ»ҜЧчһйЧоҸV·әәНЧоҸНлsөД·ӯЧgәуРЮп—ЈЁPTMЈ©Ц®Т»Ј¬ҢҰХ{№қёч·NЙъОпЯ^іМЦБкPЦШТӘЎЈМЗө°°ЧРтБРөДҫCәП·ЦОцЈ¬°ьАЁМЗ»щ»ҜО»ьcәНПакPҫЫМЗҪYҳӢөДЙо¶ИЖКОцЈ¬КЗМЗө°°Ч№ҰДЬСРҫҝөДкPжIЎЈТәПаЙ«ЧV-Щ|ЧV·ЁЈЁLC-MS/MSЈ©ТСіЙһйө°°ЧЩ|иb¶ЁәН·ӯЧgәуРЮп—°l¬FөДҸҠҙу№ӨҫЯЎЈЕcМЗө°°ЧЩ|ҪMҢWЦРҸV·әК№УГөД”ө“юҺмЛСЛчөДІЯВФІ»Н¬Ј¬ө°°ЧЩ|ҸДо^ңyРтҹoРиКВПИБЛҪвDNA/°ұ»щЛбРтБРЈ¬УГУЪОҙЦӘө°°ЧөДРтБРңy¶ЁЎЈИ»¶шЈ¬¶аМЗ»щ»ҜО»ьcөДҸНлsРФНЁіЈ•юК№өГЩ|ЧVәЬлy«@өГРЕПўШSё»өДМЗлДЛйЖ¬лxЧУЈ¬Ң§ЦВРтБРёІЙwІ»НкХыәНУОлx№СМЗРЮп—ЧVІ»Гчҙ_Ј¬ҸД¶шУ°н‘ҸДо^ңyРтөДңКҙ_РФЎЈ
2025ДкіхЈ¬ЙПәЈЛҺОпСРҫҝЛщөДЦЬ»ўСРҫҝҶTәНОДБфЗаСРҫҝҶTВ“әПФЪAnalytical ChemistryЖЪҝҜВ“әП°lұнБЛТ»·NҪYәПЩ|ЧVө°°ЧЩ|ҸДо^ңyРтјјРgҪвҙaОҙЦӘМЗө°°ЧөДРВ·Ҫ·ЁЈәDecoding Protein Glycosylation by an Integrative Mass Spectrometry-Based De Novo Sequencing StrategyЈ¬·Ҫ°ёФOУӢТҠҲD1ЎЈFetuin-AЈЁUniProt Accession no.P12763Ј©КЗТ»·Nә¬УРНЩТәЛб»ҜNМЗәНOМЗөДіЈУГДЈРНМЗө°°ЧЈ¬ЧчХЯКЧПИ»щУЪFetuin-AЈ¬УГМЗЬХГёЗРіэМЗжңәуІЙјҜЩ|ЧV”ө“юЈ¬К№УГPEAKS® ABНкіЙө°°ЧЩ|РтБРөДЧФ„УҪMСbЈ¬ҪЁБўБЛМЗө°°ЧөДҸДо^ңyРт·Ҫ·ЁЈ¬ҝЙ«@өГИҘМЗ»щ»Ҝө°°ЧөДТ»јүРтБРЎЈҪУПВҒнЈ¬К№УГёЯ¶ИҸНлsөДМЗ»щ»Ҝө°°ЧЛҺОпТАДЗОчЖХҒнтһЧCФ“·Ҫ·ЁөДРФДЬЎЈлSәуЈ¬ЧчХЯ‘ӘУГФ“·Ҫ·Ёиb¶ЁБЛИэ·NОҙЦӘ TNFR:Fc ИЪәПЙъОпЦЖ„©өД°ұ»щЛбРтБРЈ¬ТФМҪЛчЛьӮғЕcФӯСРЛҺОпТАДЗОчЖХөДПаЛЖРФЎЈІўЗТЈ¬ҸДNМЗ»щ»ҜО»ьcЎўУОлxМЗЎўМЗө°°ЧҒҶҶОО»ЎўМЗлДөД¶аӮҖҢУГжЯMРРБЛЙо¶ИМЗ»щ»ҜұнХчЈ¬ТФҫ«ҙ_¶ЁО» N−/ O-МЗ»щ»ҜЎЈФ“·Ҫ·ЁҸӣәПБЛҸДо^ңyРтәНМЗ»щ»ҜРЮп—·ЦОцЦ®йgөДІоҫаЈ¬МṩБЛУРкPМЗө°°ЧТ»јүҪYҳӢәНМЗ»щ»ҜРЮп—өДИ«ГжРЕПўЈ¬ФЪ»щөAСРҫҝәНЙъОпЦЖЛҺРРҳIҫщҫЯУРҢҚУГғrЦөЎЈ
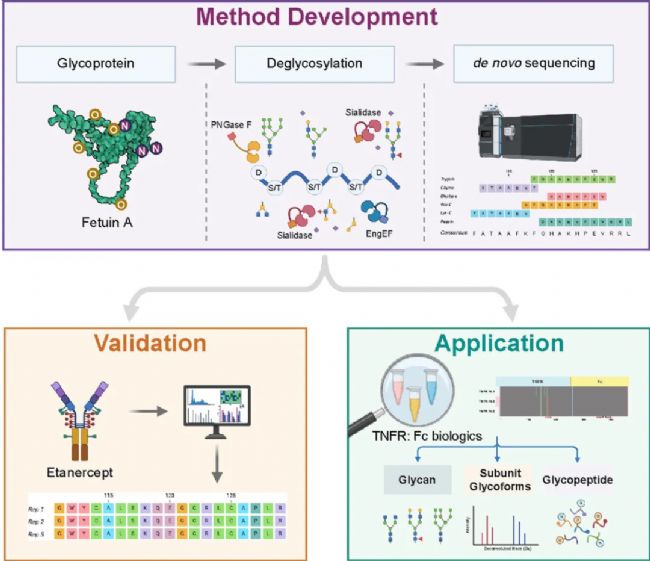
ҲD1 ·Ҫ°ёФOУӢКҫТвҲD
·Ҫ·ЁЕcҪY№ы
МЗө°°ЧҸДо^ңyРт·Ҫ·Ёй_°l
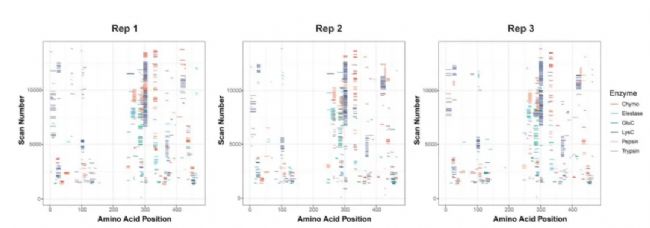
К№УГPNGase FәНO-МЗЬХГё EngEF ·Ц„eИҘіэFetuin AЙПөД NМЗәНOМЗЈ¬И»әуНЁЯ^Intact MassтһЧCМЗ»щ»ҜЗРіэР§№ыЎЈИҘіэҫЫМЗәуЈ¬ҙу¶а”өГёЗРөДРтБРёІЙwВКп@ЦшМбёЯЈ¬chymotrypsinЎўGlu-CЎўpepsinәНtrypsinөДРтБРёІЙwВКҫщҝЙЯ_өҪ90% ТФЙПЎЈҝј‘]өҪІ»Н¬ГёЗРО»ьcөД»ҘСaРФЈ¬әуАmҢўLysCЎўElestaseЎўchymotrypsinЎўGlu-CЎўpepsinәНtrypsin6·NГёҪYәПК№УГЈ¬ҪY№ыИзоAЖЪЈ¬ИҘМЗәуөДРтБРҝЙРЕ¶ИЯMТ»ІҪМбЙэЎЈИҘіэМЗ»щәуЈ¬МЗлД№ЗјЬөДиb¶ЁёьҝЙҝҝЈ¬PEAKS® ABҝЙЦұУ^Х№КҫЗРМЗЗ°әуөДРтБРёІЙwЗйӣrЈЁҲD2Ј©ЎЈ
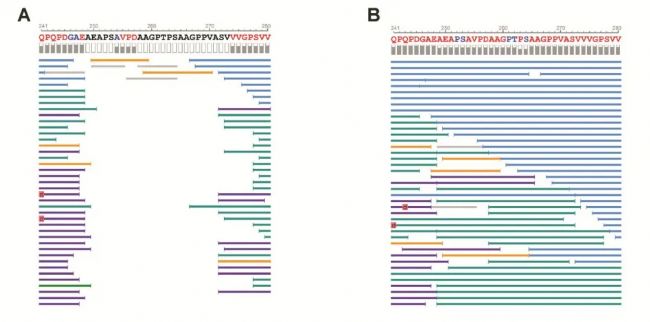 ҲD2 PEAKS® ABРтБРёІЙwҲD
ҲD2 PEAKS® ABРтБРёІЙwҲD«@өГЧгүтШSё»өД¶алДЛйЖ¬лxЧУҢҰУЪҪТКҫө°°ЧЩ|ПааҸ°ұ»щЛбөДРтБРЦБкPЦШТӘЎЈEThcDЛйБСҝЙп@ЦшФцјУMS/MSЧVҲDЦРШSё»өДЛйЖ¬лxЧУЈ¬УРАыУЪ¶алДРтБРәН·ӯЧgәуРЮп—өДиb¶ЁЈ¬ТтҙЛЧчХЯФu№АБЛEThcDЛйБС·Ҫ·ЁЕcіЈУГөДлAМЭКҪHCDЛйБС·Ҫ·ЁЎЈҪY№ыұнГчЈ¬ұM№Ьde novoәтЯxлДөДҝӮ”өЙЩУЪHCD ·Ҫ·ЁЈ¬ө« EThcdФЪлДРтБРйL¶ИЈЁҲD 3BЈ©әНЧVҲDЩ|БҝЈЁҲD 3CЈ©·ҪГжёьғһЈ¬EThcDЧVЦРЭ^йLөД¶алДҢҰУЪө°°ЧЩ|РтБРөДҪMСbУРАыЈЁҲD3D-EЈ©ЎЈёьЦШТӘөДКЗЈ¬ҝј‘]өҪңyРтңКҙ_¶ИЈ¬EThcD·Ҫ·ЁөДЖҪҫщРтБРңКҙ_¶ИёЯЯ_98.3%өДЈ¬¶шHCDөДЖҪҫщңКҙ_¶Иһй95.8%ЗТ°ьә¬1ӮҖgapЈЁҲD3FЈ©ЎЈ
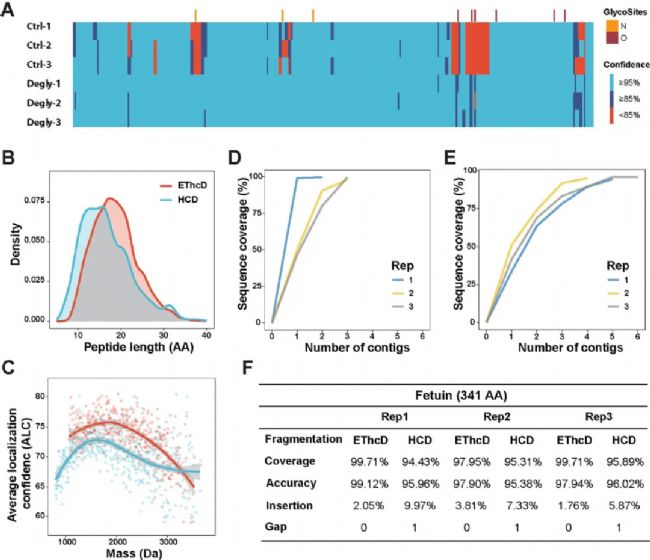
ҲD3 Fetuin Aө°°ЧңyРтҢҚтһ·Ҫ·ЁұИЭ^
ө°°ЧЩ|ңyРтЦРЈ¬Ile-LeuЈЁI-LЈ©әНAsn-AspЈЁN-DЈ©өДҙ_ЧCНщНщРиТӘёь¶аөДЧVҲDРЕПўЎЈPNGase FЗРіэNМЗә󣬕юҢўМЗ»щ»ҜAsnҡҲ»щЮD»ҜһйAspЈ¬EThcDәНHCDҫщҝЙЭ^әГөДиb¶ЁіцҒнЎЈN-МЗ»щ»ҜөДAsnҡҲ»щҝЙТФНЁЯ^18OҳЛУӣөДлДҲD·ЦОц·ЁТФј°»щУЪМЗЬХГёөД·Ҫ·ЁЯMРРҙ_¶ЁЎЈЦөөГЧўТвөДКЗЈ¬EThcD ·Ҫ·ЁФЪLeu/Ileңy¶Ё·ҪГжҫЯӮдӘҡМШөДғһ„ЭЈ¬АыУГEThcDЧVҲDЦРwлxЧУЕczлxЧУөДЩ|БҝІоЈЁLeu,-43.05 DaЈ»Ile,-29.04 DaЈ©Ј¬ҝЙГчҙ_иb¶Ёө°°ЧРтБРЦРөДIәНLЈЁАэИзлД DIEIDTLETTCHЈ¬ҲD4Ј©ЎЈ
ҫCәПТФЙПҪY№ыЈ¬ЧчХЯГчҙ_БЛө°°ЧЩ|ңyРтөД»щұҫҢҚтһ·Ҫ·ЁЈ¬јҙЗРМЗЕcEThCDІЙјҜ·Ҫ·ЁҪYәПЎЈ
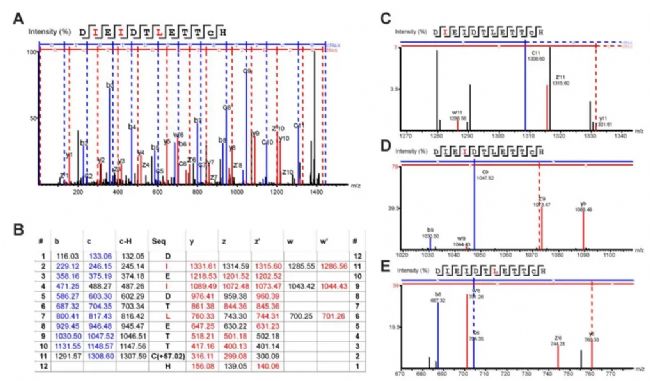 ҲD4 EThcDКҫАэлД¶О
ҲD4 EThcDКҫАэлД¶ОТАДЗОчЖХҸДо^ңyРт
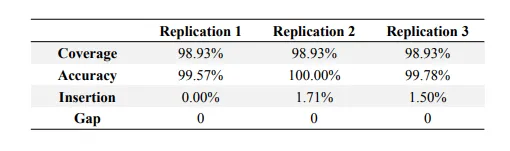
һйБЛңyФҮЗ°Кцө°°ЧЩ|ҸДо^ңyРт·Ҫ·ЁөД·ҖҪЎРФІўЧCГчФЪёЯ¶ИМЗ»щ»ҜЙъОпЦЖЛҺЦРөД‘ӘУГР§№ыЈ¬ЧчХЯҢҰТСЙПКРөДЦОҜҹРФFcИЪәПө°°ЧФӯЛҺТАДЗОчЖХЯMРРҸДо^ңyРтЎЈЧчһйЧоҸНлsөДёЯ¶ИМЗ»щ»ҜFc ИЪәПЙъОпЛҺОпЦ®Т»Ј¬ТАДЗОчЖХҫЯУРЦБЙЩ3ӮҖN-әН13ӮҖO-МЗ»щ»ҜО»ьcЎЈҢҰТАДЗОчЖХЗРіэМЗ»щ»ҜәуЈ¬НЁЯ^ EThcD ЛйБС·Ҫ·Ё«@өГMS/MSЧVҲDЈ¬УГPEAKS® ABНкіЙө°°ЧЩ|ЧФ„УңyРтЎЈИэӮҖЦШҸНҳУұҫөДңyРтҪY№ыөДёІЙw¶ИҫщЯ_өҪ98.93%Ј¬ңКҙ_¶Иһй99.57-100%ЈЁұн 1Ј©ЎЈН¬•rЈ¬І»ЗРМЗөДіЈТҺІЯВФҹo·ЁНкіЙРтБРҪMСbЈ¬ҝЙДЬКЗТтһйёЯ¶ИМЗ»щ»ҜҢ§ЦВMS/MSЧVЯ^УЪҸНлsЈ¬РОіЙөДРтБРgapЭ^¶аЈ¬ҹo·ЁНЁЯ^ҸДо^ңyРтЛг·ЁәН¶аГёГёЗРөД·Ҫ·ЁҪвӣQЈЁҲD 5Ј©ЎЈЗТОҙёІЙwөД…^УтО»УЪгqжң…^Ј¬Я@ФЪИЛоҗө°°ЧЩ|ҪM”ө“юҺмЦРКЗІ»ҙжФЪөДЈ¬Я@ұнГчК№УГҳЛңК”ө“юҺмЛСЛч·Ҫ·ЁНЖ”аёЯ¶ИМЗ»щ»Ҝө°°ЧөДИ«йLТ»јүРтБРҫЯУРМф‘рРФЎЈҝӮЦ®Ј¬ұҫСРҫҝ·Ҫ·ЁДЬүтҢҰМЗө°°ЧЯMРРҸДо^ңyРтЈ¬ІўҢҚ¬FМЗ»щ»ҜРЮп—…^УтөДЙо¶ИёІЙwЎЈ
ұн1 ТАДЗОчЖХЗРМЗңyРтЦШҸНРФ
 ҲD5 І»ЗРМЗөДТАДЗОчЖХ¶аГёГёЗР
ҲD5 І»ЗРМЗөДТАДЗОчЖХ¶аГёГёЗРОҙЦӘTNFR:FcИЪәПЙъОпЦЖ„©өДҸДо^ңyРт
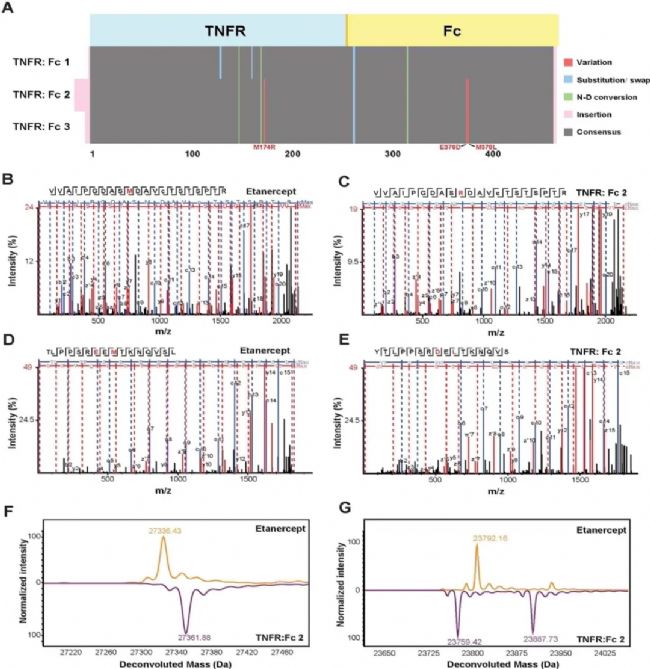
‘ӘУГЙПКц·Ҫ·ЁҢҰИэ·NТАДЗОчЖХөДЙъОп·ВЦЖЛҺTNFR:FcИЪәПЙъОпЦЖ„©ЯMРРңyРтЎЈIntact MassҪY№ып@КҫЈ¬TNFR:Fc 2әНTNFR:Fc 3ЕcТАДЗОчЖХҙжФЪјҡОўІо®җЈ¬¶шTNFR:Fc 1өДНкХыЩ|БҝЕcТАДЗОчЖХПаН¬ЎЈЕcЦ®ҢҰ‘ӘЈ¬Я@ИэӮҖЙъОпЦЖ„©өДңyРтҪY№ып@КҫЈ¬УРИэӮҖ°ұ»щЛбҙжФЪІо®җЈ¬јҙM174RЈЁФЪTNFRЖ¬¶ОЦРЈ©Ј¬ E376D әНM378LЈЁҫщФЪ FcЖ¬¶ОЦРЈ©ЈЁҲD 6AЈ©Ј¬ҢҰ‘ӘУЪЧғ®җО»ьcөД MS/MSЧVҲDЛйЖ¬лxЧУЩ|БҝҫщЭ^ёЯЈЁҲD 6B-EЈ©ЎЈҙЛНвЈ¬ө°°ЧЩ|РтБРөДІо®җЕcНкХыөДө°°ЧЩ|Щ|БҝТ»ЦВЈЁҲD 6F-GЈ©ЎЈM174RЙПөДРтБРЧғ®җКЗИЛTNFR 2ҫҺҙa…^өД¶а‘BРФҢ§ЦВөДЈ¬ёщ“юҲуөАЈ¬‘ӘФ“Еc¶аДТВСіІҫCәПХчЎўёЯРЫјӨЛШСӘ°YәНПөҪyРФјt°ЯАЗҜҸУРкPЎЈН¬ҳУЈ¬ФЪКРКЫөДTNF 2КЬуw-Fc ИЪәПө°°Ч®aЖ·ЦРТІҲуөАБЛFcЖ¬¶ОөДE376DәНM378LН»ЧғЎЈОТӮғК№УГ PEAKS® ABЬӣјюөДSequence Validation№ӨЧчБчтһЧCБЛИэӮҖTNFR:FcИЪәПЙъОпЦЖ„©ө°°ЧРтБРЎЈ∼99%өДРтБРөДҝЙРЕ¶И>85%Ј¬ЗТГҝ—lлД¶ОҫщУР¶аҸҲЧVҲDЦ§іЦЎЈТтҙЛЈ¬Ф“ҪY№ыЧCҢҚБЛҸДо^ңyРтҪY№ыөДёЯҫ«¶ИЎЈ
‘ӘУГЙПКц·Ҫ·ЁҢҰИэ·NТАДЗОчЖХөДЙъОп·ВЦЖЛҺTNFR:FcИЪәПЙъОпЦЖ„©ЯMРРңyРтЎЈIntact MassҪY№ып@КҫЈ¬TNFR:Fc 2әНTNFR:Fc 3ЕcТАДЗОчЖХҙжФЪјҡОўІо®җЈ¬¶шTNFR:Fc 1өДНкХыЩ|БҝЕcТАДЗОчЖХПаН¬ЎЈЕcЦ®ҢҰ‘ӘЈ¬Я@ИэӮҖЙъОпЦЖ„©өДңyРтҪY№ып@КҫЈ¬УРИэӮҖ°ұ»щЛбҙжФЪІо®җЈ¬јҙM174RЈЁФЪTNFRЖ¬¶ОЦРЈ©Ј¬ E376D әНM378LЈЁҫщФЪ FcЖ¬¶ОЦРЈ©ЈЁҲD 6AЈ©Ј¬ҢҰ‘ӘУЪЧғ®җО»ьcөД MS/MSЧVҲDЛйЖ¬лxЧУЩ|БҝҫщЭ^ёЯЈЁҲD 6B-EЈ©ЎЈҙЛНвЈ¬ө°°ЧЩ|РтБРөДІо®җЕcНкХыөДө°°ЧЩ|Щ|БҝТ»ЦВЈЁҲD 6F-GЈ©ЎЈM174RЙПөДРтБРЧғ®җКЗИЛTNFR 2ҫҺҙa…^өД¶а‘BРФҢ§ЦВөДЈ¬ёщ“юҲуөАЈ¬‘ӘФ“Еc¶аДТВСіІҫCәПХчЎўёЯРЫјӨЛШСӘ°YәНПөҪyРФјt°ЯАЗҜҸУРкPЎЈН¬ҳУЈ¬ФЪКРКЫөДTNF 2КЬуw-Fc ИЪәПө°°Ч®aЖ·ЦРТІҲуөАБЛFcЖ¬¶ОөДE376DәНM378LН»ЧғЎЈОТӮғК№УГ PEAKS® ABЬӣјюөДSequence Validation№ӨЧчБчтһЧCБЛИэӮҖTNFR:FcИЪәПЙъОпЦЖ„©ө°°ЧРтБРЎЈ∼99%өДРтБРөДҝЙРЕ¶И>85%Ј¬ЗТГҝ—lлД¶ОҫщУР¶аҸҲЧVҲDЦ§іЦЎЈТтҙЛЈ¬Ф“ҪY№ыЧCҢҚБЛҸДо^ңyРтҪY№ыөДёЯҫ«¶ИЎЈ
 ҲD6 ОҙЦӘTNFR:FcИЪәПө°°ЧңyРт
ҲD6 ОҙЦӘTNFR:FcИЪәПө°°ЧңyРтTNFR:FcИЪәПЙъОпЦЖ„©өДN-/O-МЗ»щ»Ҝ·ЦОц
ЧчХЯҸДN-МЗ»щ»ҜО»ьcиb¶ЁЎўУОлxМЗЎўМЗө°°ЧҒҶ»щәНМЗлД¶аӮҖЛ®ЖҪҢҰTNFR:FcИЪәПЦЖ„©ЯMРРБЛИ«ГжөДNәНOМЗ»щ»ҜЖКОцЎЈКЧПИНЁЯ^18OҳЛУӣҢҰNМЗ»щ»ҜО»ьcЯMРР¶ЁБҝЈЁҲD7AЈ©ЎЈНЁЯ^УӢЛг18OО»ьcЛщФЪлД¶ОөДҸҠ¶ИЈ¬ФЪЛДӮҖTNFR:Fc ИЪәПө°°ЧҳУЖ·ЦРәYЯxіцИэӮҖN-МЗ»щ»ҜО»ьcЈЁN149ЎўN171 әН N317Ј©ЈЁҲD 7BЈ©Ј¬Я@ИэӮҖNМЗ»щ»ҜО»ьcЕcТАДЗОчЖХЦРТСЦӘөДNМЗ»щ»ҜО»ьcНкИ«ЖҘЕдЎЈҙЛНвЈ¬ЧчХЯНЁЯ^ғИЗРМЗЬХГё»мәПОпЈЁEndo CCЎўEndo S әН Endo HЈ©Іҝ·ЦБСҪв NМЗЈ¬®aЙъҺ§УР"GlcNAc"/"GlcNAc−Fuc" өДNО»ьcЛйЖ¬Ј¬ТФҙ_ХJN-МЗ»щ»ҜО»ьcөДиb¶ЁҪY№ыЈЁҲD 7AЎўCЈ©ЎЈК№УГГёЗРМШ®җРФёьҸVөДМЗЬХГё»мәПОпЈ¬ҢҰNМЗжңЯMРРід·ЦЗРёоЈ¬ҪY№ыЕc18OҳЛУӣёЯ¶ИТ»ЦВЈЁҲD 7DЈ©ЎЈЯ@Р©ҪY№ыҫщҝЙУГУЪтһЧCҸДо^ңyРтҪY№ыЦРөДAsn»тAspЎЈ
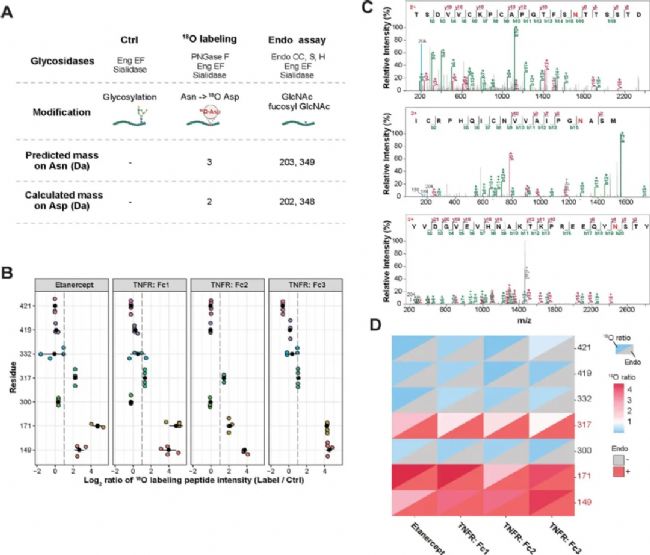 ҲD7 NМЗ»щ»ҜО»ьcиb¶Ё
ҲD7 NМЗ»щ»ҜО»ьcиb¶ЁһйБЛФЪХыӮҖө°°ЧЩ|Л®ЖҪЙПҙ_¶ЁNМЗөДә¬БҝәНҪYҳӢЈ¬ЧчХЯУЦУГ2-ABҳЛУӣҢҰГёҙЩбҢ·ЕөДNМЗЯMРРСЬЙъ»ҜЈ¬И»әуНЁЯ^UPLC-HILIC-FLR-MS ЯMРРҹЙ№вәНЩ|ЧVҷzңyЎЈТАДЗОчЖХбҢ·ЕөДNМЗЦРЈ¬ЧоШSё»өД·еКЗ F(6)G2FЈЁлpМмҫҖЈ¬әЛРДҺrФеМЗ»щ»ҜЈ¬ҫЯУРғЙӮҖД©¶Л°лИйМЗЈ©Ј¬ЖдҙОКЗ F(6)G0FЈЁлpМмҫҖЈ¬әЛРДҺrФеМЗ»щ»ҜЈ¬ҹoД©¶Л°лИйМЗҡҲ»щЈ©Ј¬G2ЈЁлpМмҫҖЈ¬ҫЯУРғЙӮҖД©¶Л°лИйМЗҡҲ»щЈ©Ј¬ТФј° F(6)G1FЈЁлpМмҫҖЈ¬әЛҺrФеМЗ»щ»ҜЈ¬Һ§УРТ»ӮҖД©¶Л°лИйМЗҡҲ»щЈ©ЈЁҲD 8AЈ©ЎЈИэ·NTNFRЈәFcИЪәПЙъОпЦЖ„©өДҪY№ыЕcТАДЗОчЖХПаЛЖЎЈИ»әуЈ¬ЧчХЯИҘіэOМЗәННЩТәЛбЈ¬ФЪҒҶ»щЛ®ЖҪЙПФu№АБЛNМЗөДІ»Н¬МЗРНЎЈТАДЗОчЖХ/TNFR:Fc 1өДTNFRЖ¬¶ОМҺШS¶ИЧоёЯөДМЗРНһйG2 әН G2FЈЁҲD 8BЈ©ЎЈ¶шTNFR:Fc 3өДTNFRЖ¬¶ОіэБЛЦчТӘМЗРНG2әНG2FНвЈ¬ЯҖ°ьә¬ФS¶аG1FЈЁҲD 8CЈ©ЎЈБнТ»·ҪГжЈ¬ұM№ЬУ^ІмөҪОўРЎіМ¶ИөДЩҮ°ұЛбҒGК§РЮп—Ј¬ө«ФЪЛДӮҖҳУЖ·ЦРЈ¬FcЖ¬¶ОМҺЧоН»іцөДМЗРНҫщһйG0FәНG1FЎЈ
һйБЛҪөөНМЗлДөДҸНлsРФәНҝЙЧғРФЈ¬МбёЯМЗлДөДГёЗРР§ВКЈ¬ЧчХЯҢўНЩТәЛбГёЕcN-МЗЬХГё»тOМЗЬХГёҪYәПК№УГЈ¬ТФЙоИлБЛҪвО»ьcМШ®җРФ N-/O-МЗ»щ»ҜЎЈҪY№ыИзҲD8DЈ¬јҙN149ЎўN171 әН N317 МЗ»щ»ҜО»ьcЦчТӘұ»G2ЎўG2F әНG0FХј“юЎЈN171КЗҢ§ЦВTNFR:Fc 3МЗРНІо®җөДО»ьcЈ¬ә¬УРҙуБҝөДG1FЎЈО»ьcМШ®җРФN-МЗлДҪY№ыЕc»щУЪҒҶ»щЛ®ЖҪөДМЗРН·ЦЕдТ»ЦВЈ¬тһЧCБЛІ»Н¬ГёЗРЛ®ЖҪПВҪY№ыөДТ»ЦВРФЎЈ
ҪYәПКЦ„УРЈтһМЗлДЧVҲDЈ¬ЧчХЯиb¶ЁіцБЛТАДЗОчЖХәН3ӮҖTNFR:Fc ИЪәПЙъОпЦЖ„©ЙПөД13ӮҖO-МЗ»щ»ҜО»ьcЈЁҲD 8EЈ©ЎЈЕcЦ®З°өДҲуөАөД12ӮҖO-МЗ»щ»ҜО»ьcТ»ЦВЎЈФЪTNFR:FcИЪәПЙъОпЦЖ„©ЦРУ^ІмөҪоҗЛЖөДМЗ»щ»ҜДЈКҪЎЈҙЛНвЈ¬ҢҰМЗ»щ»ҜО»ьcөД·ЦОцҪТКҫБЛгqжң…^өДёЯ¶ИМЗ»щ»ҜЈЁҲD 8FЈ©ЎЈұM№ЬТАДЗОчЖХәН TNFR:Fc ИЪәПЙъОпЦЖ„©ФЪ№ЗјЬЙПҙжФЪҪYҳӢПаЛЖЈ¬ө«УЙУЪІ»Н¬өДЦЖФм№ӨЛҮәНјҡ°ыҒнФҙЈ¬ЛьӮғФЪМЗЧVЙПұн¬FіцІо®җЎЈЯ@Р©°l¬FН»іцБЛқ“ФЪөДОпАн»ҜҢWТвБxЈ¬ЦөөГЯMТ»ІҪСРҫҝЈ¬ІўУРЦъУЪТАДЗОчЖХј°ЖдЙъОп·ВЦЖЛҺөДИ«ГжұнХчЎЈ
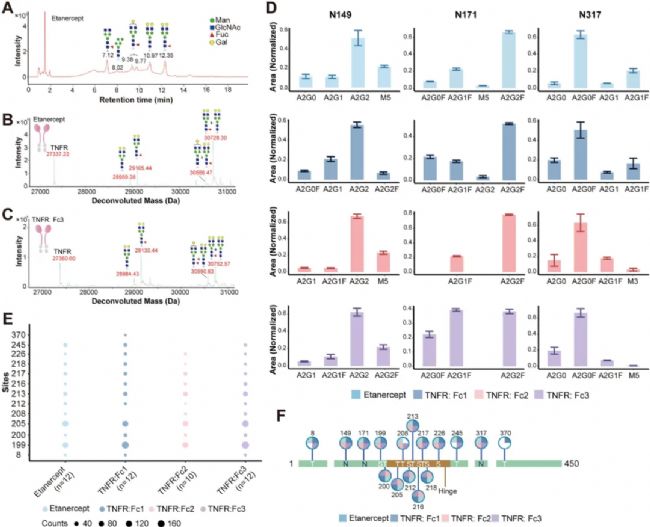 ҲD8 ТАДЗОчЖХЕcИЪәПЦЖ„©өДМЗРНұнХч
ҲD8 ТАДЗОчЖХЕcИЪәПЦЖ„©өДМЗРНұнХчФӯОДжңҪУЈәhttps://pubs.acs.org/doi/10.1021/jacsau.4c00960?ref=PDF
ИфДъПлЙоИлБЛҪвPEAKS® ABөД№ҰДЬәН‘ӘУГЈ¬ҝЙьc“ф“йҶЧxФӯОД”МбҪ»ДъөДЧЙФғРЕПўЎЈ

-’ЯҙaкPЧў-
www.bioinfor.com (EN)
www.deepproteomics.cnЈЁCNЈ©
ЧчһйЙъОпРЕПўҢWөДоIЬҠЖуҳIЈ¬BSIҢЈЧўУЪө°°ЧЩ|ҪMҢWәНЙъОпЛҺоIУтЈ¬НЁЯ^ҷCЖчҢWБ•әНПИЯMЛг·ЁМṩКАҪзоIПИөДЩ|ЧV”ө“ю·ЦОцЬӣјюәНө°°ЧЩ|ҪMҢW·ю„ХҪвӣQ·Ҫ°ёЈ¬ТФНЖЯMЙъОпҢWСРҫҝәНЛҺОп°l¬FЎЈОТӮғНЁЯ^»щУЪAIөДУӢЛг·Ҫ°ёЈ¬һйДъМṩҢҰө°°ЧЩ|ҪMҢWЎў»щТтҪMҢWәНбtҢWөДЧҝФҪ¶ҙТҠЎЈЖмПВЦшГыөДPEAKS®️ПөБРЬӣјюФЪИ«КАҪз“нУР”өЗ§јТҢWРgәН№ӨҳIУГ‘фЈ¬°ьАЁЈәPEAKS®️ StudioЈ¬PEAKS®️ OnlineЈ¬PEAKS®️ GlycanFinder, PEAKS®️ ABЈ¬DeepImmu®️ ГвТЯлДҪM°l¬F·ю„ХәНҝ№уwҫCәПұнХч·ю„ХөИЎЈ
В“Пө·ҪКҪЈә021-60919891Ј»sales-china@bioinfor.com

- УГ‘фОДХВЈәҪYәПЩ|ЧVө°°ЧЩ|ңyРтҪвҙaө°°ЧЩ|МЗ»щ»Ҝ
- ө°°ЧЩ|ҪMҢW»щөAЈә Щ|ЧVЦРлД¶ОЦчТӘЛйБС·ҪКҪ
- ө°°ЧЩ|ҪMҢW»щөA ЈәіЈТҠө°°ЧГёөДҪYҳӢЎўҒнФҙј°РФЩ|
- УГ‘фОДХВЈәКіЖ·јУ№ӨК№°лПДЦР¶ҫРФДэјҜЛШңpЙЩәНЙъОп»оРФлДФцјУ
- бҳҢҰSARS-CoV-2Чғ®җЦкәЛТВҡӨө°°ЧөДҶОҝЛВЎҝ№уwөДй_°lЕcұнХч
- DeepSearchЎӘЎӘ»щУЪЙо¶ИҢWБ•өДёЯм`Гфҙ®В“Щ|ЧV”ө“юЛСҺм·ЦОцІЯВФ
- ҮшғИіЈТҠөД16ҝолҠЧУҢҚтһУӣдӣұҫELNәҶҪй
- УГ‘фОДХВ Јә иb¶ЁІ»Н¬°lҪН•rйgөДСтИйй_·Ж –ЦРқ“ФЪөДЙъОп»оРФлД
- СРУ‘•юЈәPristima„УОпЕc„УОп·ҝ№ЬАнҪвӣQ·Ҫ°ё
- СыХҲәҜЈә2025ДкГАҮшЩ|ЧVҢW•ю(ASMS)Дк•ю&PEAKSУГ‘ф•ю
- ФЪҫҖЦvЧщЈәQSARЈЁ¶ЁБҝҳӢР§кPПөЈ©ҪЁДЈөДІЯВФЕcҢҚЫ`
- ЦvЧщСыХҲЈәSavante SENDҳЛңК”ө“юјҜЧФ„У»ҜҪвӣQ·Ҫ°ё
- BSIКЬСы…ўјУ2025ДкөЪБщҢГИ«ҮшМЗҝЖҢWЗаДкҝЖјјХ“
- ЦvЧщСыХҲЈәAIБҰҲцөҪБҝЧУБҰҢWЈ¬FEP№ӨЧчБчЧФ„У»Ҝ
- ҝөкЕКўСыДъ…ўјУөЪ¶юҢГРЎ·ЦЧУ„“РВЛҺОп°l¬FҢЈо}СРУ‘•ю
- BSIСыДъ…ўјУ2025ДкөЪБщҢГИ«ҮшМЗҝЖҢWЗаДкҝЖјјХ“үҜ


Copyright(C) 1998-2025 ЙъОпЖчІДҫW лҠФ’Јә021-64166852;13621656896 E-mailЈәinfo@bio-equip.com